
Enfoque diagnóstico molecular utilizando secuenciación exómica en las distrofias musculares cintura-cadera
Es difícil
realizar un diagnóstico clínico debido a la heterogeneidad genética del
fenotipo de distrofias musculares cintura-cadera. La aproximación diagnóstica
es compleja y requiere clasificar las distrofias musculares según el patrón de
afectación y el patrón de herencia de la enfermedad. Adicionalmente, debido a
los múltiples genes que pueden generar clínica semejante a las diferentes
distrofias musculares, se recomienda realizar secuenciación exómica solicitando
especial énfasis en los genes candidatos de distrofias musculares
cintura-cadera.
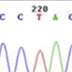
| Electroferograma de la cadena sentido con la mutación detectada por secuenciación tipo Sanger c.1130G>T en el gen LMNA. |
Secuenciación del exoma
Se
realizó secuenciación del exoma detectando variante nucleótida c.1130G>T en
el gen LMNA; esta variante produce el cambio del aminoácido arginina por
leucina en la posición 377 de la cadena polipeptídica (R377L). El resultado fue
confirmado posteriormente mediante secuenciación Sanger; en la Figura 2 se
observa un cambio de codón CGC por CTC con un cambio de aminoácido de arginina
por leucina en la posición 377.
En Colombia, este es el primer caso de LGMD1B
reportado y diagnosticado molecularmente, y también es la primera vez que se
reporta la mutación R377L en el gen LMNA en población latinoamericana. En el
2002, Ki et al. (14) reportaron el caso de una mujer
coreana de 41 años con contracturas en ambos tobillos, alteraciones en la
marcha y debilidad proximal de ambas extremidades, además de taquicardia atrial
con bloqueo atrioventricular de tercer grado, cardiomiopatía dilatada y
fibrilación auricular. Al secuenciar el gen LMNA se encontró la mutación R377L
y se realizó diagnóstico de LGMD1B.
Los análisis bioinformáticos
demostraron que el cambio del aminoácido arginina por leucina genera efectos
negativos con relación a la estabilidad de la proteína, generando la pérdida de
un enlace de hidrogeno entre los aminoácido 377 (L) y 374 (H). Los resultados
corroboran a nivel molecular que el SNP R377L genera efectos negativos a la
láminina A, lo que colaboraría a explicar el fenotipo de LGMD1B en la paciente.
En pacientes con clínica semejante a la
de la paciente, además de considerar el grupo de las LGMD, es fundamental tener
como diagnósticos diferenciales la EDMD2, la CMD1A y la ECMT2B1. Contrario a la
LGMD1B, la EDMD2 presenta contractura en los codos y tendón calcáneo,
restricción en los movimientos cervicales, cardiomiopatía dilatada con defectos
de conducción cardíaca y arritmias cardíacas. Además puede presentar escápula
alada, rigidez espinal, atrofia y debilidad de extremidades de predominio
superior, aumento de la creatina fosfoquinasa, compromiso muscular en la
primera década de la vida y compromiso cardíaco después de los 20 años
(5,16-20).
Análisis bioinformático del SNP R377L
Los
análisis del efecto de los polimorfismo de un solo nucleótido (SNP) R377L se
realizaron bajo condiciones ideales, con pH=7.0 y a una temperatura de 27oC. Al realizar el análisis, utilizando la estructura
cristalina de la proteína humana LMNA 1X8Y, la estabilidad de la proteína decreció
22%, lo que sugiere un aumento en la inestabilidad de la estructura
tridimensional debido al cambio del aminoácido. Igualmente, el análisis
estructural de la proteína LMNA mostró la pérdida de un enlace de hidrógeno
entre el aminoácido leucina 377 con histidina 374 posterior a la mutagénesis in silico (Figura 3) con un cambio de
la energía libre a nivel local en el cambio de aminoácido Arginina (R) por
Leucina (L) de 271KJ/mol a -35KJ/mol, y a nivel global del cristal la LMNA de
-4094KJ/mol a -3855KJ/mol (13). El análisis fue realizado con el programa
DeepView-Swiss-PdbViewer y los puentes de hidrogeno se representaron en color
verde.
 |
| Efecto de los SNP R377L sobre la formación de puentes de hidrogeno en la estructura terciaria de la proteína LMNA. |
La
CMD1A puede presentarse como cardiomiopatía congestiva, defectos en la
conducción, fibrilación o flutter auricular,
arritmia ventricular o incluso efusión pericárdica y su sintomatología
usualmente inicia alrededor de los 50 años; se diferencia de la LGMD1B ya que
esta no presenta compromiso osteomuscular (5,16). Por último, los pacientes con
ECMT2B1 presentan pie cavo, alteraciones en la marcha y debilidad en extremidades;
sin embargo, se diferencia de la LGMD1B en que su patrón de herencia es
autosómico recesivo, tiene compromiso de fuerza distal, los síntomas inician
entre los 10 y 20 años y presenta ausencia de afectación cardíaca.
Conclusiones
La secuenciación del exoma es una herramienta útil y costo-efectiva para el diagnóstico molecular de pacientes con clínica de distrofia muscular y especialmente LGMD; así mismo, es fundamental caracterizar el patrón de debilidad muscular, determinar el tipo de herencia y solicitar otros exámenes como la creatinina fosfoquinasa, la biopsia muscular y la electromiografía con velocidad de neuroconducción para poder determinar el tipo de genes y patologías que pueden generar dicho compromiso. No obstante, debido a la heterogeneidad genética de las distrofias musculares hay limitación en los diagnósticos fenotípicos y por eso se debe recurrir a técnicas moleculares para realizar un diagnóstico confirmatorio y definitivo (11); por lo tanto, los autores recomiendan la SE para este tipo de patologías como una herramienta valiosa y además sugieren la implementación del análisis in silico de las mutaciones halladas para predecir su posible patogenicidad.










No hay comentarios.: